With over half of the world’s population at risk of malaria every year, it remains one of the most important parasitic diseases and the leading cause of morbidity and mortality globally. The disease is considered endemic in 104 countries, with Africa accounting for over 90% of deaths every year. Children and pregnant women are the most affected, especially in low and middle-income countries. Over the past years, the emergence of resistance to effective antimalarial drugs, toxicity and high cost has created a high demand for the discovery of new antimalarial drugs with novel chemotypes, molecular targets and pan-activity in line with the new target product profiles (TPP) for malaria.
Our malaria research involves a hit-to-lead multi-directional approach in developing hits arising from both target-based and phenotypic screening. Different approaches are employed in the design leading up to the synthesis of both organic and organometallic target compounds. Some approaches include hybridization, repositioning, and the utilization of privileged molecular scaffolds. Our workflow also involves the interrogation of putative targets for phenotypic projects, these include targets involved in hemoglobin degradation pathway of P. falciparum, enzymology, chemoproteomics and live-cell microscopy among others. Our CADD platforms allows for rational design and synthesis of compounds targeting a diverse class of novel malaria targets. Details of our malaria research can be found in our publications [https://health.uct.ac.za/kelly-chibale-research/2020-2]
________________________________________________________________________________________________
Structure−Activity Relationship projects
Our research focusing on SAR exploration of compounds, especially heterocyclic, that showed antiplasmodium activity in High-throughput screening:
Pyrido[1,2-a]benzimidazolesas β-hematin inhibitors

Related references:
- Antimalarial Pyrido[1,2-a]benzimidazoles
- Antimalarial Pyrido[1,2-a]benzimidazoles: Lead Optimization, Parasite Life Cycle Stage Profile, Mechanistic Evaluation, Killing Kinetics, and in Vivo Oral Efficacy in a Mouse Model
- Structure–Activity Relationship Studies and Plasmodium Life Cycle Profiling Identifies Pan-Active N-Aryl-3-trifluoromethyl Pyrido[1,2-a]benzimidazoles Which Are Efficacious in an in Vivo Mouse Model of Malari
Pyrido[1,2-a]benzimidazolesas β-hematin inhibitors
Related references:
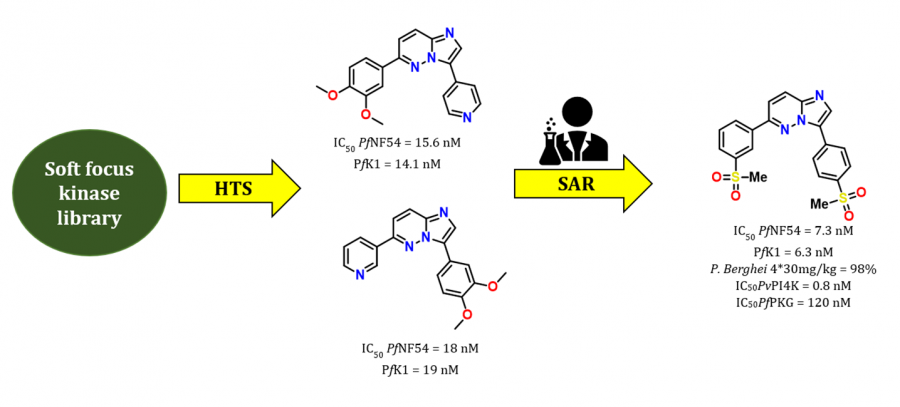
- Medicinal Chemistry Optimization of Antiplasmodial Imidazopyridazine Hits from High Throughput Screening of a SoftFocus Kinase Library: Part 1
- Medicinal Chemistry Optimization of Antiplasmodial Imidazopyridazine Hits from High Throughput Screening of a SoftFocus Kinase Library: Part 2
- New Amidated 3,6-Diphenylated Imidazopyridazines with Potent Antiplasmodium Activity Are Dual Inhibitors of Plasmodium Phosphatidylinositol-4-kinase and cGMP-Dependent Protein Kinase
- Antiplasmodial imidazopyridazines: structure–activity relationship studies lead to the identification of analogues with improved solubility and hERG profiles
________________________________________________________________________________________________
Drugs repositioning projects
The antihistamine drug Astemizole (ATS) showed growth inhibition of chloroquine-sensitive (CQ-S) and multi-drug-resistant (MDR) malaria parasites. ATS is associated with human ether-a-go-go-related gene (hERG) potassium (K+) channel inhibition. In this context, structure−activity relationship studies around AST are currently carried out within our group to overcome AST issues and to expand structure-activity relationships.

Related references:
- Structure–Activity Relationship Studies Reveal New Astemizole Analogues Active against Plasmodium falciparum In Vitro
- Multistage Antiplasmodium Activity of Astemizole Analogues and Inhibition of Hemozoin Formation as a Contributor to Their Mode of Action
________________________________________________________________________________________________
Computer-Aided drug design
The breakthrough compound MMV390048 that progressed to Phase 2 clinical trials was identified as an inhibitor of Pf phosphatidyl inositol-4 kinase (PI4K). Unlike most PfPI4K inhibitors, MMV390048 displayed remarkable selectivity towards its target over the human host kinases. The selectivity over these two analogous proteins was explained in silico by constructing a homology model and comparative molecular docking between the homology model and a Human PI4K crystal structure. These docking studies laid the groundwork for explaining selectivity patterns observed in multiple chemical series.
Details of our malaria research can be found in our publications [https://health.uct.ac.za/kelly-chibale-research/2020-2]

Related references:
________________________________________________________________________________________________
Chemical biology tools for target identification
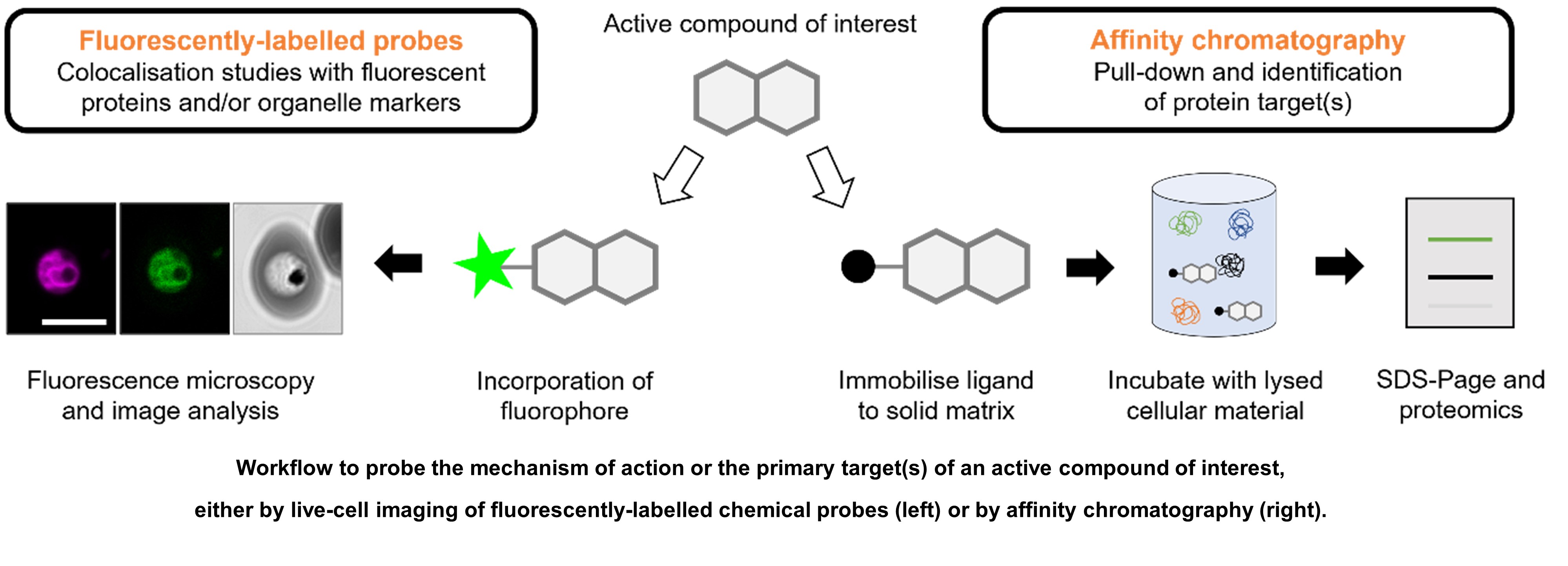
Knowing the primary target of an active compound can enable rational decision-making and accelerate drug discovery efforts. Target identification methods comprise a wide range of technologies under the umbrella of chemical biology, in which small molecule tools are used to generate valuable insights into biological processes. For example, the phenotypic effect of a compound on the morphology of cellular organelles may be observed directly via electron microscopy or with organelle-specific fluorescent dyes. Alternatively, the compound of interest may itself be covalently modified to incorporate a fluorophore. This probe may then be used to measure colocalisation with fluorescent proteins or organelle markers, revealing valuable information about the mechanism of action of the active compound.
Alternatively, affinity-based target identification methods may be used. The most common application of this approach is affinity chromatography, in which the “pull-down” of a target protein is achieved by covalently attaching the small molecule to a solid matrix such as a bead or a capturable group such as biotin. To complement this approach, we aim to establish additional label-free affinity-based assays such as the cellular thermal shift assay (CETSA) and drug affinity responsive target stability (DARTS) assay, coupled with mass spectrometry. Together, these techniques have been successfully employed for target deconvolution in the context of infectious diseases.
________________________________________________________________________________________________
Targeted covalent inhibitors
In contrast to reversible noncovalent inhibitors, targeted covalent inhibitors (TCIs) offer the potential for a new generation of antimalarial agents with improved efficacy and specificity. Covalent inhibitors comprise a reactive chemical warhead that binds to the active site of the target protein by bringing a moderately reactive electrophile in close proximity to a nucleophilic amino acid residue, typically cysteine. While there are currently over 40 FDA-approved TCIs, meaning that this modality has now been accepted as an important weapon in the chemotherapeutic arsenal, this approach has not yet been thoroughly explored in the context of malaria.
We are currently investigating Plasmodium phosphatidylinositol 4-kinase beta (PI4Kβ) TCIs specifically targeting PvPI4Kβ C1327 (PfPI4Kβ C1361). To this end, we are using a combination of in vitro PvPI4Kβ time-dependent kinase assays utilising purified recombinant protein, as well as P. falciparum whole-cell assays, both coupled with mass spectrometry, and in combination with molecular docking predictions, to guide the design of suitable covalent inhibitors.